Despite impressive success by historical standards, still the majority of patients will fail CAR T-cells. This begs the question, how do we improve the durability of response to CAR T-cells? What can we offer patients who relapse after CAR T-cells?
In designing strategies for improving the durability of CAR T-cell responses, it is helpful to first understand what limits their ability to achieve lasting remissions. When the first patients began relapsing after CAR T-cell therapy, it was tantalizing to speculate the reason for relapse would be predictable and simple, such as loss of target antigen expression. Although this is the cause of relapse in a minority of lymphoma patients, a number of additional factors are thought to contribute to the remaining relapses. These factors can be broken down into three categories, in my opinion: tumor-related, T-cell-related, and host-related:
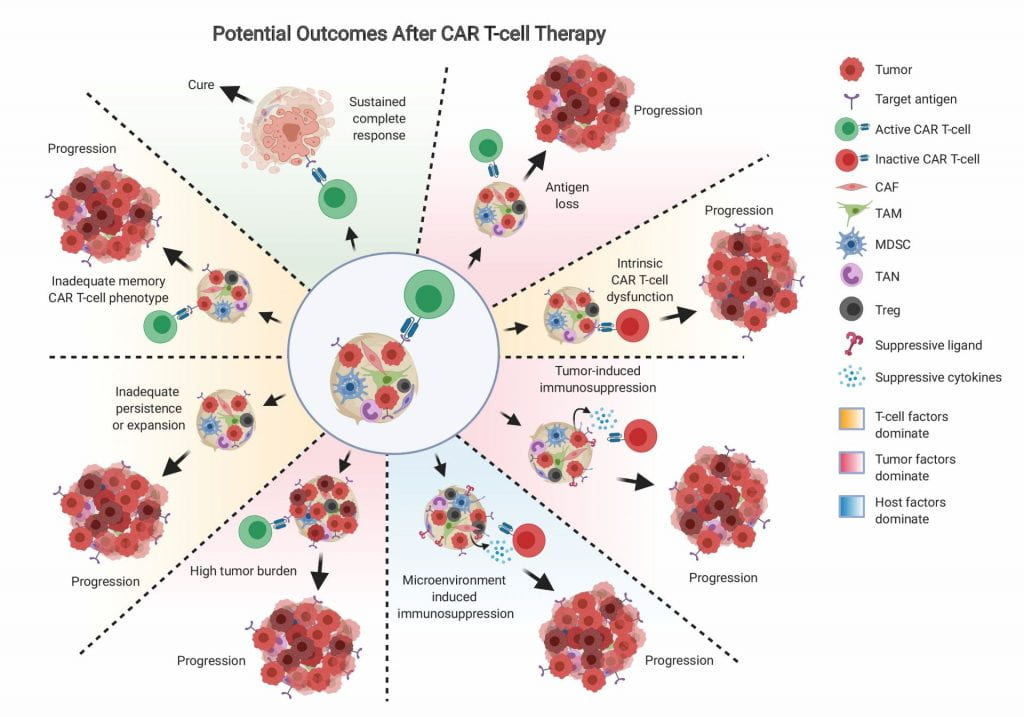
Tumor-related factors beyond loss of the target antigen include expression of inhibitory molecules such as immune checkpoint ligands, or expression of inhibitory cytokines such as TGF β or IL-10. Loss of the target antigen, specifically CD19, occurred in 27% (3 of 11 patients) of DLBCL patients treated on ZUMA-1.7 Mechanistically, antigen loss has been best studied in leukemia where it affects 10–20% of patients, and occurs through the generation of exon splice variants that eliminate the extracellular CAR binding domain, as well as tumor lineage switch to a myeloid phenotype, and outgrowth of cells with acquired CD19 mutations or loss of heterozygosity.14–18 In addition to modifying the target antigen, tumor cells can express receptors to inhibit CAR T-cell function. These immune checkpoint molecules, such as PD-1, Tim3, Lag3, and CTLA4, are best known for their function in quenching the endogenous T-cell response, although CAR T-cell specific activities have also been documented. In mice, inhibiting PD-1 pathway activation improves CAR T-cell mediated solid tumor clearance.19 A limited number of CD19 CAR-T cell-refractory DLBCL patients have been described that achieved significant CAR-T cell expansion, antitumor response, and decreased tumor burden after treatment with PD-1 blocking antibody.20–22 In an analysis performed on final CAR products going into DLBCL patients, Tim3 and Lag3 co-expression on CAR T-cells pre-infusion was found to significantly predict for negative response, suggesting that CAR T-cell exhaustion status may be largely ordained even before the cells enter the tumor.23
Since there is a finite number of tumor cells the average CAR T-cell can kill, a simple mechanism for relapse is likely the be excessive tumor burden relative to CAR T-cell number. Long-term follow-up of ALL patients treated with CAR T-cells observed a clear difference in survival between patients starting with low vs high tumor burden; in fact, the only patients who survived long-term were those who started with low tumor burden.24 In DLBCL patients treated on ZUMA-1, low baseline tumor burden also strongly predicted for durable response.25 Even among patients who achieved comparable peak CAR T-cell levels, those with higher baseline tumor burden had fewer durable responses than those with lower baseline tumor burden, and the CAR T-cell/tumor volume ratio was predictive.25
T-cell-related factors include failure to achieve adequate expansion and failure to adequately kill. These impairments can be attributed to T-cell anergy, exhaustion, or senescence. Although functionally these conditions overlap, T-cell anergy is a hyporesponsive state in T-cells which is triggered by activation of TCR signaling in the absence of adequate costimulation through CD28; T-cell exhaustion occurs when T-cells achieve adequate stimulation and costimulation, but repeated activation during chronic infection or tumor progression eventually silences their function, and T-cell senescence is characterized by growth arrest after excessive proliferation with maintained viability and metabolic activity.26 CAR T-cell exhaustion, anergy, and senescence may reflect the patient’s baseline T-cell status, and may be influenced by treatment history and the CAR manufacturing process. Like endogenous T-cells, CAR T-cells become exhausted by repetitive (tonic) signaling, which can be antigen-induced or antigen-independent.27 Over expression of the transcription factors associated with further costimulation, such as c-Jun, may ameliorate CAR T-cell exhaustion.28
The importance of CAR T-cell proliferation, which is negatively affected by anergy, exhaustion, and senescence, in achieving durable responses is well documented. CAR T-cell growth kinetics measured prior to infusion in DLBCL patients treated on ZUMA-1 showed that those with a short doubling time were significantly more likely to achieve an objective response than patients with slower CAR T-cell division in the same growth media.25 The CAR T-cell phenotype most associated with effective doubling time in this analysis was the stem-like memory cells (defined here as CCR7 +CD45RA + CD8 T-cells). Consistently, in CLL patients treated with CAR T-cells, sustained remission was associated with an elevated frequency of CD27+CD45RO–CD8+ T cells before CAR T-cell generation, and these lymphocytes possessed memory-like characteristics.29
Although long-term CAR T-cell persistence might rationally be assumed to be important to maintaining a durable response, the actual importance of CAR T-cell persistence beyond the first several months currently appears to be largely insignificant; long-term follow-up from ZUMA-1 showed that 3/4 of patients with ongoing responses had B cell recovery5 and that CAR T-cell levels at 4 weeks but not at 3 months or beyond predicted for objective and durable response25 ; data from JULIET showed no link between absolute T-cell concentration and clinical outcomes, suggesting the CAR T-cell functionality, and not the absolute numbers over time, are most meaningful.6 However, achieving a supportive environment for initial CAR T-cell engraftment, expansion and function is still critical, and currently supported by conditioning chemotherapy prior to CAR T-cell infusion.
Host factors that affect relapse after CAR T-cell therapy are largely related to the presence and dominance of immunosuppressive cells in the tumor microenvironment, such as tumor-associated macrophages (TAMs), marrow-derived suppressor cells (MDSCs), tumor-associated neutrophils (TANs), cancer-associated fibroblasts (CAFs) and regulatory T cells (Tregs). These cells are known to inhibit T-cell function in a plethora of ways, however the exact contribution of each of these cell types to CAR T-cell hyporesponsiveness remains to be studied in vivo, and may also depend on the cancer type, location, and patient.
In DLBCL patients treated with CAR T-cells in which biopsy was performed before treatment and analyzed by RNAseq, patients who went on to only achieve PR had a tumor profile upfront suggestive of more MDSCs (defined by CD33, CD14), tumor-associated fibroblasts (FAP, TNC, CSPG4, PDGFRA, S100A4, ASPN, STC1, ITGAM), and immunosuppressive cytokines (IL10, TGF-β1), compared with those who went on to achieve a complete response (CR).30 Determining the individual contribution of each of these cell types in CAR T-cell mouse models requires an immunocompetent syngeneic system with genetic knockout capacity, which has been limited primarily due to difficulty in effectively generating mouse CAR T-cells.
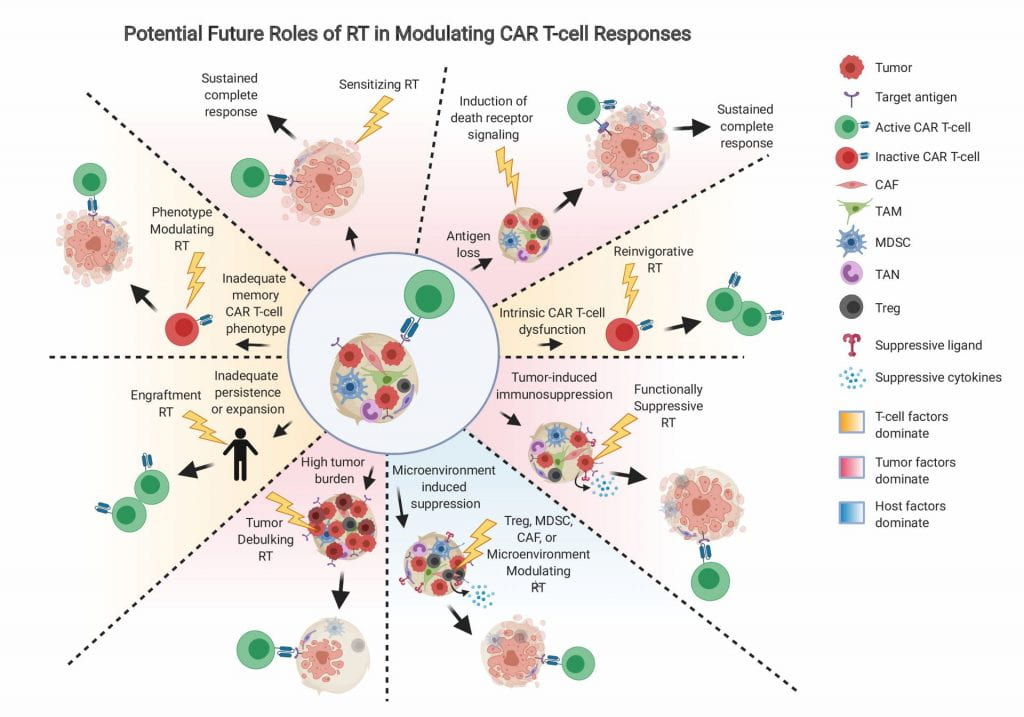
Future role of RT with CAR T-cells
As in other cancer circumstances, the current role of RT relates to addressing local disease, and is thus mostly applicable to giving a tumoricidal dose to areas that are likely to relapse after CAR T-cells, or to patients fortunate enough to have limited sites of disease prior to CAR T-cells. In the future, the role of RT may expand to include addressing the underlying mechanistic causes of CAR T-cell failure (Figure 2), which would apply to a much larger patient population.
For patients who relapse after CAR T-cells with antigen-positive disease who are eligible for retreatment with CAR T-cells, and for patients receiving CAR T-cells for the first time, neoadjuvant RT may be utilized to address the main functional limitations outlined in Figure 3. For example, RT may be utilized to specifically kill off MDSCs, Tregs, CAFs, or other immunosuppressive cells that are often rich in the tumor microenvironment.40 The precise dose of RT required to specifically ablate these cell types requires further investigation, but likely would be much less than the tumoricidal dose, and thus may be more safely deliverable to larger regions. These cells may be stained and quantified in biopsy specimens or analyzed by flow or RNAseq to predict their likely contribution to CAR T-cell resistance in advance.
While tumor-debulking RT prior to CAR T-cell infusion is appealing and should be studied more precisely on trial, disease often progresses after CAR T-cells and the dilemma of whether or not to use post-CAR RT arises. RT is generally avoided after CAR T-cell therapy for the same reason steroids were initially avoided; there is concern the therapy may impair or kill the cytotoxic cells. However, just as moderate steroid use has thus far not had a clearly inhibitory effect on CAR T-cell function in patients 32029707, it may also be true that moderate focal RT does not significantly affect CAR T-cell efficacy. For example, a multiple myeloma patient treated with 4 Gy x 5 fractions of palliative RT to the brain and spine shortly after progressing clinically and biochemically after CAR T-cells exhibited a significant expansion of T-cell clones post-RT, as well as a systemic response that was durable at last follow-up.41 It is possible that RT doses that do not kill CAR T-cells modulate their phenotype in a clinically relevant manner. It is still unclear whether the RT sensitivity of CAR T-cells is different from endogenous T-cells, and what the “CAR T-cidal” dose is. It is also unknown how lower, non-lethal doses of RT affect CAR T-cell function, specifically their tumor killing-capacity, persistence, or expansion. Tumor cells sometimes undergo a phenomenon of accelerated repopulation if they do not die after RT, and RT can independently induce inflammatory signaling in a number of cells; potentially conserved mechanisms may exist in CAR T-cells that could be harnessed to the patient’s benefit by utilizing the correct dose regimen in a form of “reinvigorative RT”.
Both CD4 and CD8 cells can develop a number of different phenotypes that subsequently affect their ability to respond to further antigen stimulation. Several studies have found associations between the memory phenotype present in patient’s CAR T-cell populations and their chance of attaining a durable tumor response.23,29,42 Thus, attention has been placed on inducing the appropriate memory T-cell phenotype in CAR-modified cells prior to infusion.43,44 Phenotype can also be plastic, and the phenotype of an injected CAR T-cell may not durably maintain in vivo over time. It is currently unknown whether low-dose RT influences CAR T-cell phenotype. In the future, if such is the case, RT may be given to patients failing CAR T-cells to re-establish a favorable phenotype and extend the potential for response.
In patients with heterogenous antigen expression, “sensitizing RT” may be utilized, which may consist of a low dose (~2 Gy) of RT to a large area to encompass all sites of disease. This approach in mice results in CAR T-cells more effectively killing antigen-positive cells, as well as nearby antigen-negative tumor cells through death receptor ligand naturally induced on activated CAR T-cells that interact with death receptor pathway molecules upregulated on and within tumor by low dose RT.37 This approach requires further validation in patients, but may be a way to improve responses in those likely to fail due to partial antigen loss.
Given the growing importance of CAR T-cell therapy and the wide array of potentially synergistic effects of RT with CAR T-cells, the next 5–10 years may be a particularly exciting time for the field of radiation oncology and cellular immune therapy. Carefully executed, well-designed clinical trials should be performed to document and test mechanistic hypothesis behind various potentially synergistic RT regimens. In doing so, RT as a localized modality may eventually achieve the elusive goal of improving cure rates of patients with metastatic disease, a goal that has thus far been unachievable.