The successful modification of T-cells with CARs to treat certain types of cancer implies a world of possibilities for future cell therapies for other indications. What will be the next major type of CAR-modified immune cell? What will be the source of those cells?
Excerpt from Carl DeSelm, Cell Types Used for CAR Generation, Springer Nature, 2021.
The most common CAR T-cell product generated and infused into patients, today and in the past, is an unsorted, αβ Τ-cell derived directly from the cancer patient. Although this personalized autologous T-cell manufacturing method has many advantages and has resulted in outstanding clinical data in hematologic malignancies, a number of aspects require improvement for cell therapy to impact broader patient populations. The first aspect relates to the poor sustained anti-tumor response of CAR T-cells in solid tumors as well as a significant group of leukemia and lymphoma patients. Utilizing cells that naturally penetrate larger tumor masses better, kill through additional mechanisms, are less prone to antigen escape or intrinsic cytotoxic resistance, or that better establish and maintain an anti-tumor microenvironment, may overcome some of the deficiencies of CAR T-cells in poorly responsive tumors. Further, utilizing other cell types may lessen the tremendous cost of autologous manufacturing, the possibility of manufacturing failure in some patients, the length of time for manufacturing, and the side effect profile. Here I will discuss alternative cellular sources developed to date, including allogeneic T-cells, Natural Killer (NK) cells, invariant NK T-cells (iNKT), induced pluripotent stem cells (iPSCs), T-cells of a defined CD4/CD8 ratio, T-cells of a defined memory phenotype, and myeloid cells modified with a CAR.
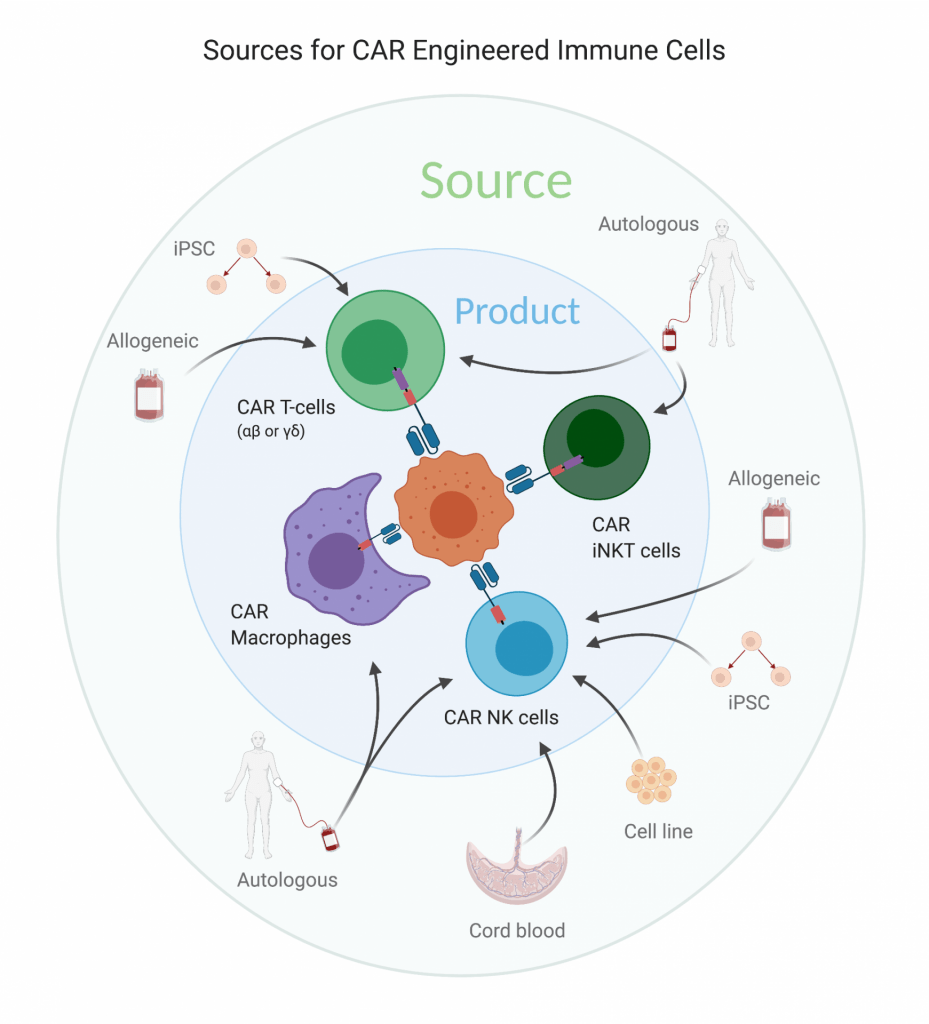
Allogeneic CAR T-Cells
For hematologic malignancies treated with CAR T-cells for which efficacy is quite impressive, a current major limitation is patient access to the product, and the financial burden placed on the healthcare system due to the high cost of manufacturing a single product from a single patient using their own autologous T-cells [1]. The current list price for the approved CAR T-cell therapies is $373,000, and the process takes roughly 3 weeks to manufacture. These 3 weeks are a vulnerable time for patients; they have already progressed on the most effective known chemotherapy regimens so keeping their disease controlled can be a challenge, and progression of disease in this time sometimes renders them ineligible to receive the manufactured CAR T-cells. Even if they remain eligible, progression in this time may make the disease less controllable by the CAR T-cells. Allogeneic, or “off-the-shelf” CAR T-cells would provide a solution to these problems. Of course, the challenging question remains, can allogeneic CAR T-cells address these limitations while still providing the same level of efficacy as autologous CAR T-cells?Theoretically, cancer patients who have anergic, exhausted, or senescent T-cells as a result of their cancer, their age, or their multiple prior lines of cytotoxic therapy [2], may enjoy improved disease response if given a CAR product from a healthy donor with optimally-functioning T-cells. However the inclination of the host immune system to reject the foreign T-cells tends to limit their efficacy, and the inclination of the foreign T-cells to reject the host’s tissues or organs (i.e., graft-versus-host disease or GVHD) introduces the potential for additional toxicities with this strategy.Overcoming these obstacles requires first understanding the mechanisms by which cells are rejected by either the host or the donor. Since GVHD is a major cause of death in allogeneic stem cell transplant, much research has been devoted to the subject, and αβ T cells have been found to be central to its pathogenesis [3]. The αβ TCR recognizes peptides presented by MHC molecules, which in humans are comprised of HLA proteins. HLA genes have more polymorphisms than any other human genes, leading to thousands of slightly different HLA protein variants. Of these many different variants, up to six MHC-I and six MHC-II variants are present in each individual. Through a process of negative selection in the thymus, T-cells are educated to not react against any self-MHC molecule, or even any self-MHC molecule loaded with a peptide derived from any of the thousands of proteins encoded in that individual’s genome. However, a new peptide presented by a self-MHC, or a different MHC variant, may still be recognized and lead to alloreactivity. Further, amino acid differences in the peptide-binding region of a given MHC molecule determine the specific sequence of peptides that are capable of being presented, and thus simply changing the MHC results in a wide variety of different peptides presented that were not present in the thymus’s negative selection process, even if the full proteins themselves are identical.While it is nearly impossible to match all HLA variants among donors and recipients, matching HLA-A, HLA-B and HLA-DR is sufficient to reduce the incidence of allogeneic graft rejection. However, establishing a matched HLA CAR T-cell program would require a large bank from a wide range of donors, and would inevitably exclude a number of patients, especially ethnic minorities or those with less common HLA variants. Further, even though partially-matched CAR T-cells may not react strongly against the host, it is likely that these infused allogeneic CAR T-cells will be more rapidly eliminated by either host T-cells or antibodies recognizing remaining discordant peptide-MHC complexes, or other surface proteins.Thus, achieving allogeneic CAR T-cell persistence is a major challenge. In addition to HLA matching, more intense lymphodepletion therapy may be required to further immunosuppress the host response against the infused CAR T-cells; however, this approach also compromises other aspects of host immunity, such as effective responses to infectious agents. Repeating intensive lymphodepletion for subsequent CAR T-cell administration carries additional risk and morbidity. A more specific, genetic approach to improving CAR T-cell persistence is to knock out (genetically delete) MHC-I, which is a potential major target of rejection of CAR T-cells by the host immune system. The most efficient way to do this is to delete β2-microglobulin, which is essential for forming functional MHC-I molecules on the cell surface. MHC-II is also expressed on activated T-cells and may also likely be a target of host immune mediated rejection of allogeneic CAR T-cells. Therefore, knocking out or blocking MHC-II on CAR T-cells would also be necessary to maximize their persistence. A relatively efficient way of doing this is to knock out the master regulator of all MHC-II molecule expression, CIITA. Knocking both MHC-I and MHC-II molecules in CAR T-cells has been achieved in mouse studies [4].Since MHC molecules are major inhibitors of NK cell cytotoxicity as they signify self, depleting MHC-I and MHC-II makes the CAR T-cells more susceptible to NK-mediated elimination. To circumvent this problem, the additional expression of non-classical HLA molecules (HLA-E or HLA-G), which can inhibit NK cells but are less common targets for T-cell rejection, can be added to the CAR vector [45].An option available to the relatively small number of patients who have previously had an allogeneic stem cell transplant and have subsequently relapsed is to generate CAR T-cells from the original stem cell donor. In this way, the host immune system, which has already been reconstituted with a different donor, will be genetically identical to the infused CAR-modified cells. In a series of 20 patients who underwent this strategy, eight had a response to CAR T-cells (six complete responses [CRs] and two partial responses [PRs]), and none developed new-onset GVHD after CAR T-cell infusion [6].In addition to solving the problem of allogeneic CAR T-cell persistence, the problem of GVHD needs to be addressed. Beyond HLA matching, preventing GVHD can be successfully achieved through a variety of other means, such as knocking out the TCR from the infused cells, using a cell product with a restricted TCR profile (such as only those that recognize a viral antigen), or using cell types that naturally lack an αβ TCR. The TCR is a heterogeneous group of proteins consisting of either an α-chain and a β-chain (in αβ Τ cells) or a γ-chain and a δ-chain (in γδ T cells), as well as four separate CD3 transmembrane proteins (CD3δ, CD3γ, CD3ε and CD3ζ). The β-chain contains two possible constant regions, while the α-chain has one, making it logistically easier to abolish the αβ TCR by targeting the single α-chain (TRAC). Methods have progressed from successfully knocking out the TRAC in CAR T-cells [7], to knocking the CAR gene into the TRAC locus, putting the CAR under the natural transcriptional regulation of the TCRα and ameliorating some of the exhaustive effects of tonic signaling from constitutive high CAR expression [8]. TCR knockout CAR T-cells (UCART19) have shown feasibility in two clinical trials with relapsed B-cell leukemia, demonstrating a 67% CR, and a 6-month PFS of 27% [9]. In these studies, the UCART19 product has both the TCR and the mature lymphocyte marker CD52 knocked out, which allows for additional lymphodepletion but not CAR T-cell depletion using the monoclonal anti-CD52 antibody alemtuzumab. This strategy thus both prevents the CAR T-cells from recognizing the host through its TCR (deleted by CRISPR/Cas9), and reduces the host’s ability to eliminate the CAR T-cells by depleting host lymphocytes (via infusion of alemtuzumab). How this approach will compare to autologous CAR T-cell efficacy remains to be seen in further studies.While knocking out the TCR is an eloquent approach, it requires sophisticated and relatively expensive techniques. Another potential strategy, which still utilizes intact αβ T-cells, is to selectively employ memory T-cells that recognize a viral antigen for CAR transduction. Since the risk of GVHD is proportional to the diversity of the TCRs present, selecting for a smaller number of TCR clones against a known, non-human target should greatly reduce the risk. However, since TCRs are degenerate, the possibility remains that a particular antiviral TCR may still cross-react with a host tissue antigen. Additionally, since these are all T-cells that have previously encountered the viral antigen at least once, the baseline phenotype will be different; the effect of using this population of prior antigen-exposed cells is unknown. This approach has been demonstrated in glioblastoma patients targeting HER2 in a clinical trial, which showed feasibility and one partial response among 17 patients [9]. Whether this approach is will be effective against a more responsive tumor like ALL remains to be reported in clinical trials.
Macrophages
Macrophages, being completely unrelated to T-cells on the hematologic family tree, have a plethora of different functions that make them attractive candidates for CAR modification in cases where T-cells encounter changes. For example, while T-cells poorly infiltrate solid tumors, macrophages are often summoned to them by cytokines released by the tumor; the actively migrate into the tumor against a pressure gradient while T-cells largely lack this capacity. While T-cells kill primarily through inducing apoptosis by granzyme and perforin secretion, to which tumor cells have variably levels of innate and acquired resistance, macrophages can theoretically dispose of their targets by direct phagocytosis. Additionally, they have been known to exert cytotoxic effects through antibody dependent cytotoxicity (ADCC). Unfortunately, macrophages are usually drawn to the tumor microenvironment to provide a “healing” role; they sense destruction and arrive to reduce inflammation and promote recovery. This role generally has the effect of promoting tumor growth, spread, and metastasis. Since they are highly plastic, they can change from this reparative, pro-tumor role to an inflammatory, anti-tumor role and back again, depending on their stimuli.Engineering macrophages with a CAR is an appealing way to genetically instruct them to maintain an inflammatory anti-tumor phenotype, as well as to infiltrate and phagocytose tumor. Macrophages modified with a CAR containing a signaling domain from Megf10 or FcRɣ have both been found to achieve target-specific phagocytosis [33]. A CAR macrophage targeting the extracellular matrix protein CD147 successfully reduced tumor collagen deposition and improved T-cell infiltration [34]. CAR macrophages expressing a CD3z-based CAR phagocytosed tumor in vitro and reduced tumor burden in vivo, expressed proinflammatory cytokines and chemokines, converted bystander M2 macrophages to M1, resisted the effects of immunosuppressive cytokines, and activated anti-tumor T-cells [35]. Challenges to macrophage therapy include genetically modifying the cells in large numbers, and potentially finding ways to increase the magnitude of their direct anti-tumor efficacy in vivo, as studies thus far have shown tumor reduction but not elimination. Given the number of T-cell supportive functions CAR macrophages have, they may function synergistically with CAR T-cells; however, these studies have yet to be reported. One Phase I clinical trial is currently open utilizing CAR macrophages for HER2 expressing tumors (NCT04660929).
Neutrophils
Neutrophils, like macrophages, often have pro-tumor functions, however they are relatively less well characterized in the tumor microenvironment. Their ability to kill through alternative mechanisms, such as netosis, is conceptually appealing. Before CAR T-cells had gained significant momentum, a report in 1998 of neutrophils being modified with a CAR (then called a CIR, for chimeric immune receptor) containing a CD3z intracellular domain showed antigen-specific tumor lysis [36]. However, no CAR neutrophil reports have been generated since. Whether these cells may someday play a more central role in CAR therapies remains to be seen.
iPSC
iPSCAn attractive source for theoretically unlimited CAR T-cells, or other immune cells, for allogeneic use is iPSCs. These cells could be genetically modified in numerous ways over time, unlike primary cells which are limited by the number of possible transductions before the must be used. For example, theoretically the TCR could be knocked out to prevent GVHD, MHC-I and MHC-II genes could be deleted to prevent T-cell rejection, non-classical HLA molecules (such as HLA-E or HLA-G) could be introduced to prevent NK cell rejection, additional inhibitory molecules such as PD1 could be removed, and/or they could be made to express stimulatory cytokines. Alternatively, a broad repository of HLA typed iPSCs could be utilized to create HLA-matched infusion products. Even without knocking out the TCR, since one clone can be used to generate all of the cells, the risk of GVHD would likely be very low. However, drawbacks of this approach are that iPSCs are difficult and expensive to culture in great quantities, and the process of differentiation into fully functional effector immune cells such as T-cells or NK cells after modification with a CAR is not a trivial process. However, iPSCs have been successfully used to generate CAR NK cells with functionality similar to CAR T-cells and less signs of GVHD in mice [25]. Human clinical trials with iPSC derived CARs are ongoing with results yet to be reported.
References:
1. Lin JK, Muffly LS, Spinner MA, Barnes JI, Owens DK, Goldhaber-Fiebert JD. Cost Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Multiply Relapsed or Refractory Adult Large B-Cell Lymphoma. J Clin Oncol 2019;37:2105-19.
2. Thommen DS, Schumacher TN. T Cell Dysfunction in Cancer. Cancer Cell 2018;33:547-62.
3. Zeiser R, Blazar BR. Acute Graft-versus-Host Disease – Biologic Process, Prevention, and Therapy. N Engl J Med 2017;377:2167-79.
4. Kagoya Y, Guo T, Yeung B, et al. Genetic Ablation of HLA Class I, Class II, and the T-cell Receptor Enables Allogeneic T Cells to Be Used for Adoptive T-cell Therapy. Cancer Immunol Res 2020;8:926-36.
5. Gornalusse GG, Hirata RK, Funk SE, et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol 2017;35:765-72.
6. Brudno JN, Somerville RP, Shi V, et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J Clin Oncol 2016;34:1112-21.
7. Torikai H, Reik A, Liu PQ, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 2012;119:5697-705.
8. Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017;543:113-7.
9. Benjamin R, Graham C, Yallop D, et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1 studies. Lancet 2020;396:1885-94.
10. Kato Y, Tanaka Y, Miyagawa F, Yamashita S, Minato N. Targeting of tumor cells for human gammadelta T cells by nonpeptide antigens. J Immunol 2001;167:5092-8.
11. Capsomidis A, Benthall G, Van Acker HH, et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol Ther 2018;26:354-65.
12. Tang X, Yang L, Li Z, et al. Erratum: First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res 2018;8:1899.
13. Liu E, Tong Y, Dotti G, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018;32:520-31.
14. Liu E, Marin D, Banerjee P, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med 2020;382:545-53.
15. Wagner JA, Berrien-Elliott MM, Rosario M, et al. Cytokine-Induced Memory-Like Differentiation Enhances Unlicensed Natural Killer Cell Antileukemia and FcgammaRIIIa-Triggered Responses. Biol Blood Marrow Transplant 2017;23:398-404.
16. Gang M, Marin ND, Wong P, et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood 2020;136:2308-18.
17. Xu Y, Liu Q, Zhong M, et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J Hematol Oncol 2019;12:49.
18. Chaidos A, Patterson S, Szydlo R, et al. Graft invariant natural killer T-cell dose predicts risk of acute graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Blood 2012;119:5030-6.
19. Leveson-Gower DB, Olson JA, Sega EI, et al. Low doses of natural killer T cells provide protection from acute graft-versus-host disease via an IL-4-dependent mechanism. Blood 2011;117:3220-9.
20. Rubio MT, Bouillie M, Bouazza N, et al. Pre-transplant donor CD4(-) invariant NKT cell expansion capacity predicts the occurrence of acute graft-versus-host disease. Leukemia 2017;31:903-12.
21. Schneidawind D, Pierini A, Alvarez M, et al. CD4+ invariant natural killer T cells protect from murine GVHD lethality through expansion of donor CD4+CD25+FoxP3+ regulatory T cells. Blood 2014;124:3320-8.
22. Rotolo A, Caputo VS, Holubova M, et al. Enhanced Anti-lymphoma Activity of CAR19-iNKT Cells Underpinned by Dual CD19 and CD1d Targeting. Cancer Cell 2018;34:596-610 e11.
23. Gottschalk C, Mettke E, Kurts C. The Role of Invariant Natural Killer T Cells in Dendritic Cell Licensing, Cross-Priming, and Memory CD8(+) T Cell Generation. Front Immunol 2015;6:379.
24. Heczey A, Liu D, Tian G, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014;124:2824-33.
25. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018;23:181-92 e5.
26. Gattinoni L, Lugli E, Ji Y, et al. A human memory T cell subset with stem cell-like properties. Nat Med 2011;17:1290-7.
27. Sommermeyer D, Hudecek M, Kosasih PL, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016;30:492-500.
28. Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 2016;126:2123-38.
29. Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 2020;396:839-52.
30. Turtle CJ, Hanafi LA, Berger C, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 2016;8:355ra116.
31. Deng Q, Han G, Puebla-Osorio N, et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat Med 2020;26:1878-87.
32. Fraietta JA, Lacey SF, Orlando EJ, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 2018;24:563-71.
33. Morrissey MA, Williamson AP, Steinbach AM, et al. Chimeric antigen receptors that trigger phagocytosis. Elife 2018;7.
34. Zhang W, Liu L, Su H, et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br J Cancer 2019;121:837-45.
35. Klichinsky M, Ruella M, Shestova O, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol 2020;38:947-53.
36. Roberts MR, Cooke KS, Tran AC, et al. Antigen-specific cytolysis by neutrophils and NK cells expressing chimeric immune receptors bearing zeta or gamma signaling domains. J Immunol 1998;161:375-84.